Anderson Lab
Mutations and Evolution
The cells in our body are constantly buffeted by exogenous agents that damage our DNA, like cigarette smoke and UV radiation. Molecular mechanisms work to quickly repair damage, but these can lead to mutations that are evolutionary capital, endowing cells with the qualities to become cancer.
Many chemotherapeutics also work by damaging DNA, so understanding the repair mechanisms at work is vital so that the disease can be treated effectively and resistance avoided. A recent surprise in our understanding of this process came from the discovery that DNA damage is tolerated, and is capable of being inherited as cells divide.
A latent mutagenic potential therefore exists every time a damaged strand acts as a template for replication, but how this is modulated is unknown. I hypothesise that the tolerance and build-up of heritable DNA damage in our genomes has a multi-faceted role in the generation of mutations and that uncovering this will provide new insights into disease progression.

Patterns of mutations can show the lingering, latent mutagenitcity of DNA damage
What we do
In my lab at the German Cancer Consortium (DKTK) site in Munich, I combine fundamental laboratory experiments, high throughput sequencing methods and advanced analytical techniques to interrogate the mutagenic mechanisms driving cancer evolution.
Principally, I want to know what modulates the patterns of mutations from tolerated DNA damage and to understand how this changes over the course of our lives.
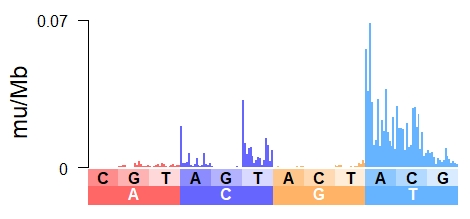
Mutation signatures reveal biases in DNA damage, repair and tolerance
Lab members
-

Craig Anderson
Junior Group Leader
-

Katrin Köllisch
PhD Student
-

You?
Biologists, geneticists, mathematicians and medics who share our research passions…
Get in touch!
Selected Publications
-
https://doi.org/10.1101/2025.01.13.632787
Human cancers are heterogeneous. Their genomes evolve from genetically diverse germlines in complex and dynamic environments, including exposure to potential carcinogens. This heterogeneity of humans, our environmental exposures, and subsequent tumours makes it challenging to understand the extent to which cancer evolution is predictable. Addressing this limitation, we re-ran early tumour evolution hundreds of times in diverse, inbred mouse strains, capturing genetic variation comparable to and beyond that found in human populations. The sex, environment, and carcinogenic exposures were all controlled and tumours comprehensively profiled with whole genome and transcriptome sequencing. Within a strain, there was a high degree of consistency in the mutational landscape, a limited range of driver mutations, and all strains converged on the acquisition of a MAPK activating mutation with similar transcriptional disruption of that pathway. Despite these similarities in the phenotypic state of tumours, different strains took markedly divergent paths to reach that state. This included pronounced biases in the precise driver mutations, the strain specific occurrence of whole genome duplication, and differences in subclonal selection that reflected both cancer susceptibility and tumour growth rate. These results show that interactions between the germline genome and the environment are highly deterministic for the trajectory of tumour genome evolution, and even modest genetic divergence can substantially alter selection pressures during cancer development, influencing both cancer risk and the biology of the tumour that develops. -
https://doi.org/10.1038/s41586-024-07490-1
DNA base damage is a major source of oncogenic mutations. Such damage can produce strand-phased mutation patterns and multiallelic variation through the process of lesion segregation. Here we exploited these properties to reveal how strand-asymmetric processes, such as replication and transcription, shape DNA damage and repair. Despite distinct mechanisms of leading and lagging strand replication, we observe identical fidelity and damage tolerance for both strands. For small alkylation adducts of DNA, our results support a model in which the same translesion polymerase is recruited on-the-fly to both replication strands, starkly contrasting the strand asymmetric tolerance of bulky UV-induced adducts. The accumulation of multiple distinct mutations at the site of persistent lesions provides the means to quantify the relative efficiency of repair processes genome wide and at single-base resolution. At multiple scales, we show DNA damage-induced mutations are largely shaped by the influence of DNA accessibility on repair efficiency, rather than gradients of DNA damage. Finally, we reveal specific genomic conditions that can actively drive oncogenic mutagenesis by corrupting the fidelity of nucleotide excision repair. These results provide insight into how strand-asymmetric mechanisms underlie the formation, tolerance and repair of DNA damage, thereby shaping cancer genome evolution.
-
https://doi.org/10.1073/pnas.2403871121
DNA base damage is a major source of oncogenic mutations and disruption to gene expression. The stalling of RNA polymerase II (RNAP) at sites of DNA damage and the subsequent triggering of repair processes have major roles in shaping the genome-wide distribution of mutations, clearing barriers to transcription, and minimizing the production of miscoded gene products. Despite its importance for genetic integrity, key mechanistic features of this transcription-coupled repair (TCR) process are controversial or unknown. Here, we exploited a well-powered in vivo mammalian model system to explore the mechanistic properties and parameters of TCR for alkylation damage at fine spatial resolution and with discrimination of the damaged DNA strand. For rigorous interpretation, a generalizable mathematical model of DNA damage and TCR was developed. Fitting experimental data to the model and simulation revealed that RNA polymerases frequently bypass lesions without triggering repair, indicating that small alkylation adducts are unlikely to be an efficient barrier to gene expression. Following a burst of damage, the efficiency of transcription-coupled repair gradually decays through gene bodies with implications for the occurrence and accurate inference of driver mutations in cancer. The reinitation of transcription from the repair site is not a general feature of transcription-coupled repair, and the observed data is consistent with reinitiation never taking place. Collectively, these results reveal how the directional but stochastic activity of TCR shapes the distribution of mutations following DNA damage.
-
https://doi.org/10.1038/s41586-020-2435-1
Cancers arise through the acquisition of oncogenic mutations and grow by clonal expansion. Here we reveal that most mutagenic DNA lesions are not resolved into a mutated DNA base pair within a single cell cycle. Instead, DNA lesions segregate, unrepaired, into daughter cells for multiple cell generations, resulting in the chromosome-scale phasing of subsequent mutations. We characterize this process in mutagen-induced mouse liver tumours and show that DNA replication across persisting lesions can produce multiple alternative alleles in successive cell divisions, thereby generating both multiallelic and combinatorial genetic diversity. The phasing of lesions enables accurate measurement of strand-biased repair processes, quantification of oncogenic selection and fine mapping of sister-chromatid-exchange events. Finally, we demonstrate that lesion segregation is a unifying property of exogenous mutagens, including UV light and chemotherapy agents in human cells and tumours, which has profound implications for the evolution and adaptation of cancer genomes.
-
https://doi.org/10.1073/pnas.1718831115
As corresponding author
Within the mega-pest lineage of heliothine moths are a number of polyphagous, highly mobile species for which the exchange of adaptive traits through hybridization would affect their properties as pests. The recent invasion of South America by one of the most significant agricultural pests, Helicoverpa armigera, raises concerns for the formation of novel combinations of adaptive genes following hybridization with the closely related Helicoverpa zea. To investigate the propensity for hybridization within the genus Helicoverpa, we carried out whole-genome resequencing of samples from six species, focusing in particular upon H. armigera population structure and its relationship with H. zea. We show that both H. armigera subspecies have greater genetic diversity and effective population sizes than do the other species. We find no signals for gene flow among the six species, other than between H. armigera and H. zea, with nine Brazilian individuals proving to be hybrids of those two species. Eight had largely H. armigera genomes with some introgressed DNA from H. zea scattered throughout. The ninth resembled an F1 hybrid but with stretches of homozygosity for each parental species that reflect previous hybridization. Regions homozygous for H. armigera-derived DNA in this individual included one containing a gustatory receptor and esterase genes previously associated with host range, while another encoded a cytochrome P450 that confers insecticide resistance. Our data point toward the emergence of novel hybrid ecotypes and highlight the importance of monitoring H. armigera genotypes as they spread through the Americas.